



NEWS




.jpg)






.jpg)


























.jpg)

.jpg)

.jpg)

.jpg)




(1).jpg)


.jpg)
.jpg)


.jpg)



.jpg)

























































Themes
Tags
- Annual Convention of the APPT
- Fundraising
- Government of Spain
- Infants
- VI Congreso Internacional en torno a la Acondroplasia y Otras Displasias Óseas
- 0-3 years
- 3-6 years
- 6-12 years
- Adults
- ALPE Camp
- Ascendis Pharma
- BMN 111-206
- Book
- Carnival
- Child
- Committee on the Rights of Persons with Disabilities
- Courses
- Covid-19
- Education
- Family
- Infigratinib
- Institut Imagine
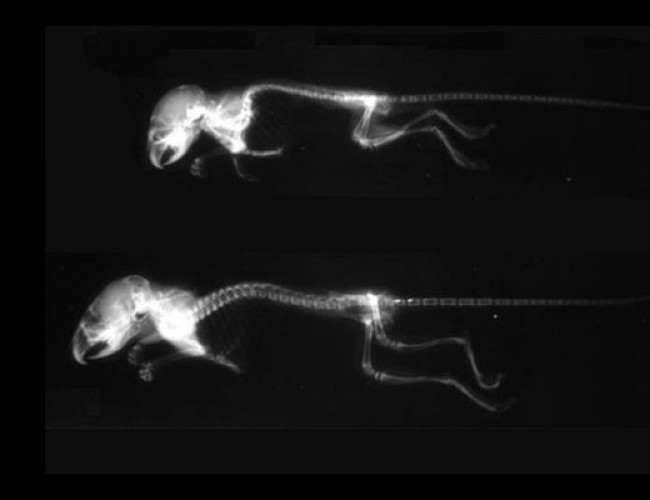
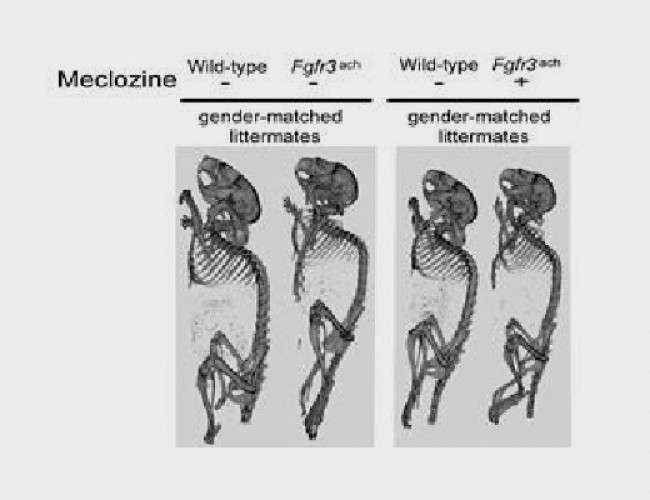
- Meclizina
- Meclozine
- Nutrition
- Pharmacotherapy
- Programa de Atención Integral a las personas con ADEE y sus familias
- Psychology
- Publications
- QED Therapeutics
- Reportage
- Solidarity initiative
- Therachon
- UN Recommendations
- Universidad de Nagoya
- Vosoritide
- World Orphan Drug Congress WODC 2018
ALPE Achondroplasia Foundation was created on January 24, 2000, thanks to the enthusiasm of several persons, primarily Carmen Alonso, Miguel López, and the Press-Lewis family. The Press-Lewis family had founded ProChon Biotech Ltd. in Tel-Aviv (Israel) to find a therapy for achondroplasia. ProChon was the origin of scientific advances in the investigation of achondroplasia, which continues to provide more and more interesting results





MEDICAL SERVICES
Fundación ALPE Acondroplasia
Calle Conde Real Agrado, 2
33205 Gijón